{kind=link}

Imaging Transwells with Zeiss 710 MP microscope
In publications with data from this microscope, please acknowledge grant NCRR S10 RR023704-01A1.
Cells were put in Costar Transwell Polycarbonate Membrane Permeable Supports EF5650C and EF5650B. Either they were prelabeled or stained post-fixation with fluorescent dye. Each well was removed from the original chamber and placed in a MatTek dish P35G-1.5-14-C on the microscope stage for imaging.
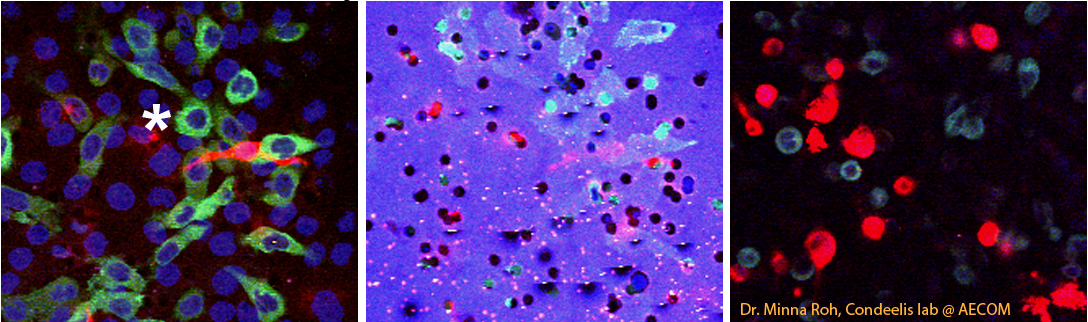
First tries were with standard confocal microscopy with the 63X N.A. 1.4 objective. We were able to image fluorescence of the cells and reflectance of the filters simultaneously. [example image]
After discussion with Minna Roh in the Condeelis lab at AECOM, who sent impressive images, we decided to switch to multi-photon. We switched to a 40X water immersion objective. We had a problem imaging the filter, however. Whereas Roh showed that the filters she used emitted light across the visible spectrum when hit with the pulsed IR laser, we did not detect this in any channels available (420 to 760 nm detectors). Therefore, we decided to sequentially image fluorescence of cells by 2-photon and reflection by 488 nm.
|
We have been using the 40X (metal top) N.A. 1.1 Water LD C-APOCHROMAT Korr UV-VIS-IR SN 421867-9970 with Immersol W as a water substitute. |
Example image after Z axis adjustment: mum17sideslices_mp_example_1.jpg
The second example shows (on the far right) the relationship of the cells and and membrane before and after reregistration in the Z axis. mum17sideslices_mp_example_2.jpg
This is caused by chromatic aberration. The reflection is at 488 nm and the multiphoton excitation is at 840 nm. Based on measurements with beads, the focal point from one to the other wavelength shifts by approximately 3 um.
  |
The images above (montage at the left) show single sections of a Z series taken at 1 um steps with the 40X water objective of green fluorescent beads glued to a MatTek dish and in water. These settings are the same as for the cells in the transwells. The top row is excitation of green fluorescent beads by 2-photon at 840 nm. The middle row is excitation of the beads by 1-photon 488 nm. The bottom row is reflection imaging at 488 nm. The montage at the right illustrates the shift by reslicing in the XZ axis.
Also, the light paths of the external and internal detectors are not in register in XY. Therefore, a simple XY translation needs to be applied to the images to accurately show the spatial relationship of the cells to the holes in the filter. These macros multiphoton_and_standard_confocal_registration_v100.txt are a quick, if not super user friendly way, to register all axes.
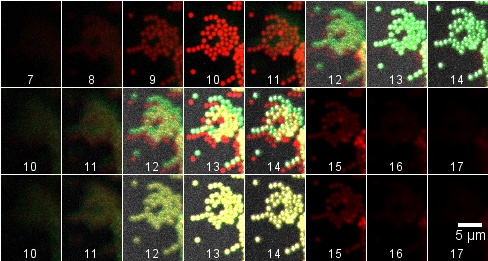
Top row: raw data Z stacks merged.
Middle row: 3 um inserted at beginning of red (840 nm) stack and 3 um at the end of the green stacks. (The last three frames have no green in them because the original Z stack did not step far enough. It is important to make the Z series extend beyond the sample.)
Bottom row: Red stack translated 4 X -6 pixels.
The registration issues may be avoided by switching the filters used for the assay to Millipore 24-well millicell hanging inserts (CAT# PIEP12R48) which are called "PET" and are designed to be more uniformly clear specifically for microscopy. These would be fluorescent in all channels (according to Roh) but could be subtracted out of the cell channel.
last updated 20110802
comments, questions, suggestions: Michael.Cammer@med.nyu.edu